Sheepy...
Colombia Médica
On-line ISSN 1657-9534 On-line ISSN 1657-9534
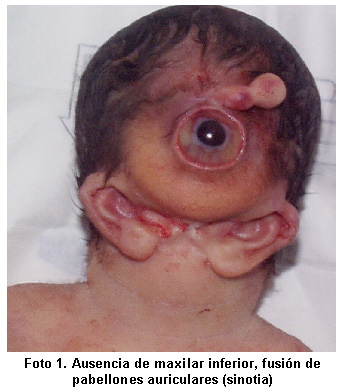
Complejo agnatia holoprosencefalia: informe de caso Agnatia holoprosencephaly complex: case report Fernando Suárez-Obando MD*, Juan Carlos Prieto, MD, M.Sc.* Fernando Suárez-Obando MD *, Juan Carlos Prieto, MD, M.Sc. * 1. Instituto de Genética Humana, Pontificia Universidad Javeriana, Bogotá, DC, Colombia. Instituto de Genética Humana, Pontificia Universidad Javeriana, Bogotá, DC Colombia. RESUMEN ABSTRACT Se presenta un caso de complejo agnatia holoprosencefalia y se realiza una revisión de la literatura, en relación con la compleja etiología genética y embriológica de este conjunto de malformaciones mayores de la cara y el sistema nervioso central. We report a case of holoprosencephaly agnatia complex and a review of literature relating to the complex genetic and embryological etiology of this set of major malformations of the face and the central nervous system. Se trata del primer caso que se informa en la literatura colombiana. This is the first case reported in the literature Colombia. Palabras clave: Genética; Holoprosencefalia; Anomalías craneofaciales. Keywords: Genetics; Holoprosencephaly; craniofacial abnormalities. Agnathia holoprosencephaly complex: case report Agnathia holoprosencephaly complex: case report SUMMARY SUMMARY A case report of the agnathia holoprosencephaly complex and a review of the literature related to the complex genetic and embryologic aetiology of this group of major birth defects of face and central nervous system are informed. A case report of the agnathia holoprosencephaly complex and a review of the literature related to the complex embryologic and genetic aetiology of this group of major birth defects of face and central nervous system are informed. The present clinical case is the first reported in Colombia. The present clinical case is the first reported in Colombia. Keywords: Genetics; Holoprosencephaly; Craniofacial anomalies. Keywords: Genetics; Holoprosencephaly; Craniofacial anomalies. El complejo agnatia holoprosencefalia, es una malformación congénita mayor, casi siempre letal, caracterizada por ausencia o hipoplasia severa del maxilar inferior, posición anormal de los pabellones auriculares y holoprosencefalia 1 . The complex agnatia holoprosencephaly, a congenital malformation is more, almost always fatal, characterized by absence or severe hypoplasia of the lower jaw, abnormal position of the ears and holoprosencephaly 1. Se presenta un caso de agnatia holoprosencefalia, asociado con ciclopía, sinoftalmia, probóscide y sinotia. We report a case of holoprosencephaly agnatia associated with ciclopía, sinoftalmia, proboscis and sinotia. La baja prevalencia de esta entidad y la extrema complejidad de la malformación dan relevancia a este informe clínico, el primero en Colombia. The low prevalence of this condition and the extreme complexity of the malformation are relevant to this clinical report, the first in Colombia. El estudio de esta anomalía sobresale en el contexto clínico y embriológico, debido a que su estudio explica las consecuencias de la falta de segmentación y desarrollo embriológico del sistema nervioso central, además de ser un cuadro clínico rico en semiología médica. The study of this anomaly in the Excel report and embryological fact that their study explains the consequences of the lack of segmentation and embryological development of the central nervous system, besides being rich in a clinical medical semiology. INFORME DE CASO CASE REPORT Recién nacido muerto de 36 semanas de edad gestacional, de madre de 28 años de edad (padres no consanguíneos), producto de segundo embarazo sin exposición a teratógenos. Newborn died 36 weeks gestational age, mother of 28 years of age (non-consanguineous parents), a result of exposure to second pregnancy without teratogenic. Control prenatal normal, hasta la semana 30, cuando a través de ecografía obstétrica se identifica, ventriculomegalia, agenesia del cuerpo calloso, posición anormal de los pabellones auriculares y retrognatia. Normal prenatal care until 30 weeks when using obstetric ultrasound is identified ventriculomegalia, agenesis of the corpus callosum, abnormal position of the ears and hypoplasia. La madre consulta por ausencia de movimientos fetales en la semana 36 de gestación, se realiza una nueva ecografía que ratifica los hallazgos anteriores y se diagnostica óbito fetal; parto por cesárea iterativa. The mother consulted by the absence of fetal movements at 36 weeks gestation, is a new ultrasound that confirmed the previous findings and is diagnosed fetal death, cesarean iteratively. Al examen físico se encuentra: Peso, 2300 g; talla, 48 cm. Physical examination found: weight, 2300 g, height 48 cm. Perímetro cefálico, 39 cm. Cephalic perimeter, 39 cm. Presencia de probóscide central, ciclopía, y ausencia de apertura bucal (astomia), agnatia (ausencia de maxilar inferior) y fusión de pabellones auriculares en la línea media o sinotia (signo clínico también denominado otocefalia) ( Foto 1 ). Presence of central proboscis, ciclopía and absence of mouth opening (astomia) agnatia (no lower jaw) and fusion of ears in the midline or sinotia (also known as clinical signs otocefalia) (picture 1). El resto del examen físico fue normal. The remaining physical examination was normal. Cariotipo de 550 bandas, bandeo G, 46, XY, sin alteraciones estructurales ni numéricas. Karyotypes of 550 bands, G-banding, 46, XY, without numerical or structural alterations. Previa firma del consentimiento informado, según se registra en la historia clínica, se llevó a cabo el estudio fotográfico y post-mortem; en éste el sistema nervioso central reveló agenesia de cuerpo calloso, sin fusión talámica, ventrículo único e hipoplasia de cerebelo confirmado una holoprosecenfalia alobar, en el macizo facial se evidenció esbozo de lengua, globo ocular único, y fusión total de nervio óptico. After signing the informed consent, as recorded in the clinical record was carried out on photographic studio and post-mortem in this central nervous system revealed agenesis of the corpus callosum, without fusion thalamus, single ventricle and hypoplastic cerebellum of a confirmed holoprosecenfalia alobar in massive facial outline was evident in language, single eyeball, and fusion of optic nerve. DISCUSIÓN DISCUSSION El complejo agnatia holoprosencefalia, o complejo disgnatia (OMIM: 202650) 2 , se caracteriza por hipoplasia severa o agenesia del maxilar inferior (agnatia) y presencia de pabellones auriculares ubicados anterior e inferiormente. The complex agnatia holoprosencephaly, or complex disgnatia (OMIM: 202650) 2, is characterized by severe hypoplasia or agenesis of the lower jaw (agnatia) and presence of ears located above and below. La gama de anormalidades asociadas incluye sinotia (fusión de los pabellones auriculares en la línea media) y la holoprosencefalia. The range of associated abnormalities including sinotia (fusion of the ears of the midline) and holoprosencephaly. El conjunto de la agnatia y sinotia se denomina otocefalia 3 . The whole of agnatia and sinotia called otocefalia 3. Una apertura bucal muy pequeña o ausente es una característica constante, incluso puede haber persistencia de la membrana bucofaríngea. A very small mouth opening or absent is a constant feature, even may have persistence of the membrane bucopharinx. Debido a la obstrucción respiratoria ya las malformaciones del sistema nervioso central, casi todos los afectados fallecen en el período perinatal. Due to respiratory obstruction and malformations of the central nervous system, almost all those affected die in the perinatal period. ETIOLOGÍA Y DESARROLLO EMBRIONARIO ETIOLOGY AND EMBRYONIC DEVELOPMENT Pauli et al. 4 , informan este complejo en dos hermanas, que fallecieron en el período perinatal, y sugieren una herencia autosómica recesiva; sin embargo, el análisis cromosómico prometafásico posterior de las dos niñas y de sus padres, estableció que el padre era portador de una translocación equilibrada y que una de las fallecidas tenía una translocación desequilibrada, siendo uno de los puntos de ruptura, el locus para la holoprosencefalia ubicado en 18p. Pauli et al. 4, report the complex in two sisters who died in the perinatal period, suggesting an autosomal recessive inheritance, but the chromosomal analysis prometafásico later the two girls and their parents, provided that the father was carrier a balanced translocation and that one of the deceased had an unbalanced translocation, being one of the points of rupture, the locus for holoprosencephaly located on 18p. Krassikoff y Sekhon 5 , comunicaron otro caso familiar donde tres afectados, eran portadores de una duplicación de 6p y monosomía de 18p. Krassikoff and Sekhon 5 reported another case in which three affected family, were carriers of a duplication of 6p and monosomy of 18p. El padre de los afectados así como su madre y hermano portaban una translocación equilibrada t (6:18). The father of those affected as well as her mother and brother carried a balanced translocation t (6:18). Los hallazgos de anormalidades cromosómicas, sugerían que la herencia no era recesiva y que el o los genes comprometidos tenían sus loci cerca de los relacionados con el locus de la holoprosencefalia, según lo insinuaba la presentación clínica. The findings of chromosomal abnormalities, suggesting that the inheritance was recessive and that the genes or their loci have committed close to the related locus of holoprosencephaly, as suggested the clinical presentation. Erlich et al. 6 observaron la transmisión de madre a hija del complejo disgnatia, pues la madre hizo una presentación clínica leve, que apuntaba a una transmisión dominante con expresividad variable. Erlich et al. 6 observed transmission from mother to daughter disgnatia complex, because the mother made a mild clinical presentation, which suggested a dominant transmission with variable expressivity. También proponían que las bases moleculares de este trastorno eran las alteraciones en el gen OTX2 (sigla en inglés para orthodenticle drosophila homolog of 2), gen homeótico ubicado en 14q21-q22, que se expresa en las regiones dorsales y ventrales del telencéfalo, diencéfalo y mesencéfalo 7 . He also suggested that the molecular basis of this disorder were alterations in the gene OTX2 (Spanish acronym for orthodenticle drosophila approval of 2), homeotic gene located on 14q21-q22, which is expressed in the dorsal and ventral regions of telencéfalo, diencephalon and mesencephalon 7. Guion-Almeida et al. 8 propusieron que los pacientes descritos por Erlich et al. 6 , en realidad sufrían el síndrome aurículo-condilar (OMIM: 602483)9, que consiste en severa micrognatia y anormalidades auriculares, sin anomalías en el sistema nervioso central. Guion-Almeida et al. 8 suggested that the patients described by Erlich et al. 6, actually suffered atrioventricular-condylar syndrome (OMIM: 602483) 9, which consists of severe micrognathia and abnormal headphones, without abnormalities in the central nervous system . Esta sugerencia no se ha podido confirmar, debido a que ambos síndromes pueden hacer parte de un mismo cuadro de la enfermedad, como lo mencionan otras descripciones clínicas en las que se ha clasificado al complejo en dos tipos: una forma severa que incluye la holoprosencefalia y otra leve en la que no hay anormalidades del sistema nervioso central (10,11). This suggestion has not been confirmed, because both syndromes can be part of a painting of the same disease as other clinical descriptions mentioned in which the compound is classified into two types: a severe form including holoprosencephaly and other slight abnormality which has no central nervous system (10,11). El modelo murino del complejo agnatia holoprosencefalia, demuestra que el fenotipo surge de mutaciones en el gen OTX2, en estado heterocigoto y que la severidad depende de otros genes modificadores en distintos loci (12), sugiriendo nuevamente un mecanismo de herencia dominante de expresividad variable. The mouse model of the complex agnatia holoprosencephaly, demonstrates that the phenotype arises from mutations in the gene OTX2 in heterozygous state and the severity depends on other modifier genes in different loci (12), again suggesting a dominant inheritance mechanism of variable expressivity. Se ha postulado que los efectos de las mutaciones del gen OTX2 y de sus genes modificadores, son la disminución de la capacidad inductiva del mesodermo precordial, que lleva a la agnatia por una migración neuronal anómala hacia las porciones ventrales del primer arco branquial y la segunda bolsa faríngea, fenómenos que suceden entre los 22 y 26 días de edad gestacional (13). It has been postulated that the effects of mutations of the gene OTX2 genes and their modifiers are the reduced ability of inductive precordial mesoderm, which leads to the agnatia by an abnormal neuronal migration toward the ventral portions of the first and second branchial arch pharyngeal pouch, phenomena that occur between 22 and 26 days gestational age (13). La expresión del gen OTX2, quizá tenga relación con la vía de señalización Sonic Hedgehog, que es una familia de proteínas de señalización intercelular que juegan un papel primordial en el desarrollo embrionario; las mutaciones en esta vía generan diversos fenotipos que incluyen la holoprosencefalia lobar, alobar y semilobar, malformaciones que pueden cursar con cebocefalia, etmocefalia o probóscide (14). OTX2 gene expression, perhaps in connection with the Sonic Hedgehog signaling pathway, which is a family of intercellular signaling proteins that play a role in embryonic development, mutations in this way generate different phenotypes including lobar holoprosencephaly, alobar and semilobar malformations that can heal without cebocefalia, or etmocefalia Proboscis (14). En el caso que aquí se informa, es obvio que la induc-ción mesodérmica se vio severamente afectada en los arcos branquiales, y que por esta razón la implantación auricular fue en la línea media (sinotia), debido a la ausencia del maxilar inferior. In the case reported here, it is obvious that the induced mesodermal-tion was severely affected in the branchial arches, and thus the handset deployment was in the midline (sinotia) due to the absence of the lower jaw. Otro aspecto llamativo del caso, es la ciclopía, que tiene la particularidad de ser un solo ojo en una sola orbita bien definida, lo que la diferencia de la ciclopía secundaria a la sinoftalmía o fusión ocular, donde las estructuras oculares y la bóveda orbital no están tan bien definidas. Another striking aspect of the case, is the ciclopía, which has the particularity of being a single eye in a single well-defined orbit, so the difference in the ciclopía secondary to ocular sinoftalmía or merger, where the ocular structures and no orbital roof are less well defined. La ciclopía o sinoftalmía, son secundarias a la pérdida de estructuras mediales de la holoprosencefalia. The ciclopía or sinoftalmía, are secondary to the loss of medial structures of holoprosencephaly. De tal modo que el defecto inicial pudo generarse en anomalías del desarrollo de las células que se derivan de las crestas neurales cefálicas por mutaciones en el gen OTX2, seguido de una inducción mesodérmica anormal en los arcos branquiales, ausencia del desarrollo en las estructuras de la línea media del cerebro secundario a falla en la estimulación del tubo neural y la consecuente ciclopía y probóscide, componentes del conjunto de anormalidades faciales de la holoprosencefalia. So that the initial defect was produced in abnormal development of cells that are derived from cephalic neural crest by OTX2 mutations in the gene, followed by an abnormal mesodermal induction in the gill arches, absence of development in the structures of the midline of the brain secondary to failure of the stimulation of the neural tube and the consequent ciclopía and proboscis, components of the facial abnormalities of holoprosencephaly. FRECUENCIA Y ASESORÍA GENÉTICA GENETICS AND FREQUENCY ADVICE Tanto la complejidad como la severidad de las malformaciones pueden explicar su baja frecuencia, debido a que la mayoría de defectos de este tipo generan la interrupción temprana del embarazo. Both the complexity and severity of malformations may explain their low frequency, because most of these types of defects generated by early termination of pregnancy. Hubo dos casos entre 200,000 nacimientos en el estudio colaborativo español de malformaciones congénitas (ECEMC); es decir, se indica una prevalencia de 1 en 100,000 casos (14), y alrededor de 80 casos se han comunicado en la literatura (15). There were two cases among 200,000 births in the Spanish collaborative study of congenital malformations (ECEMC), ie, indicated a prevalence of 1 in 100,000 cases (14), and about 80 cases have been reported in the literature (15). Este es el primer ejemplo del complejo agnatia holoprosencefalia que se informa en Colombia. This is the first example of the complex agnatia holoprosencephaly is reported in Colombia. Aunque no hay consenso absoluto sobre su patrón de herencia, el riesgo de recurrencia en una pareja completamente sana sin anomalías cromosómicas es menor de 1%; en este caso los padres no eran portadores de anormalidades cromosómicas ni afectados por holoprosencefalia lobar o semilobar. Although there is no absolute consensus on its pattern of inheritance, the risk of recurrence in a completely healthy couple with no chromosomal abnormalities are less than 1%, in this case the parents were not carriers of chromosomal abnormalities or affected by or semilobar lobar holoprosencephaly. CONCLUSIONES CONCLUSIONS El complejo agnatia holoprosencefalia constituye un grupo de malformaciones severas que compromete el desarrollo del sistema nervioso central y de los arcos branquiales; casi siempre es incompatible con la vida y su extrema complejidad puede explicar su baja frecuencia. The complex agnatia holoprosencephaly is a group of severe malformations that compromises the development of the central nervous system and the gill arches, almost always incompatible with life and its extreme complexity may explain its low frequency. REFERENCIAS REFERENCES 1 1 . . Carey J. J. Carey External ear. External ear. En: Stevenson R, Hall J, Goodman R (eds.). In: Stevenson R, Hall J, Goodman R (eds.). Human malformations and related anomalies. Human Malformations and related anomalies. New York: Oxford University Press; 1993. New York: Oxford University Press, 1993. p. p. 193-219. 193-219. | |||||||||

© 2009 Universidad del Valle, Cali, Colombia © 2009 Universidad del Valle, Cali, Colombia
Corporación Editora Médica del Valle Corporación Editora Médica del Valle
Universidad del Valle, Cali, Colombia Universidad del Valle, Cali, Colombia
Telefax: (57-2) 558-1939 Fax: (57-2) 558-1939
™Sheephogan™
Katharine Hepburn - "Life is hard. After all, it kills you."


No comments:
Post a Comment